White Paper
Converging Standards, Connecting Platforms: Achieving One Global Submission
The life sciences industry can improve the regulatory submissions process by embracing existing interoperable digital platforms and harmonized data standards.
Executive Summary
In 2024, the FDA approved 50 novel drugs, and more than two-thirds were first approved in the U.S. The same year, the EMA recommended marketing authorization for 114 drugs.
For patients, speed to market is critical. But the regulatory submissions process is becoming more complex. Innovation in the product development pipeline is matched by increasingly divergent local regulations, data requirements, and submission platforms. The submissions process is unnecessarily expensive and redundant.
The result of this complex regulatory environment is overly complex technology solutions. Simplified global submissions requirements will better enable technology to increase speed, efficiency, and compliance. For regulators, adoption of globally harmonized standards and shared digital submission platforms allows for a crucial shift away from redundant, jurisdiction-specific reviews toward live, targeted oversight — with smaller budgets.
Adoption of existing standards and platforms is less expensive, faster, and enables health authorities (HAs) to maximize limited budgets and reuse global data to get health innovations to the public more quickly.
For regulators, adoption of globally harmonized standards and shared digital submission platforms allows for a crucial shift away from redundant, jurisdiction- specific reviews toward live, targeted oversight — with smaller budgets.
Common outcomes, diverging paths
Biopharmas are investing heavily in regulatory information management (RIM) platforms. Nearly 70% of companies are expected to have an end-to-end RIM strategy in place in the near future, according to the most recent Gens Report. These investments improve data quality, operational efficiency, and speed the submission process for sponsors.
Despite these advances, the broader regulatory ecosystem remains highly fragmented, creating a complex web of submission platforms, global health authority requirements, data standards, submission pathways, and information-sharing collaborations.
As shown in Figure 1, different HAs (and even different centers within HAs) require different submission mechanisms for different regulated product types (drugs, biologics, devices, etc.) on different platforms. And, an increased focus on data-centric submissions has created an increasing number of locality-specific implementations of data standards, resulting in different requirements across HAs.
Additionally, many required data standards have been in use for more than 20 years and are outdated. These standards don’t work effectively for modern challenges (e.g., supply-chain monitoring) and don’t meet the need for better digital information for public use. HAs are slow to adopt new standards and routinely implement their own specific versions, adding to implementation timelines and global complexity, and reducing efficiency and speed to market.
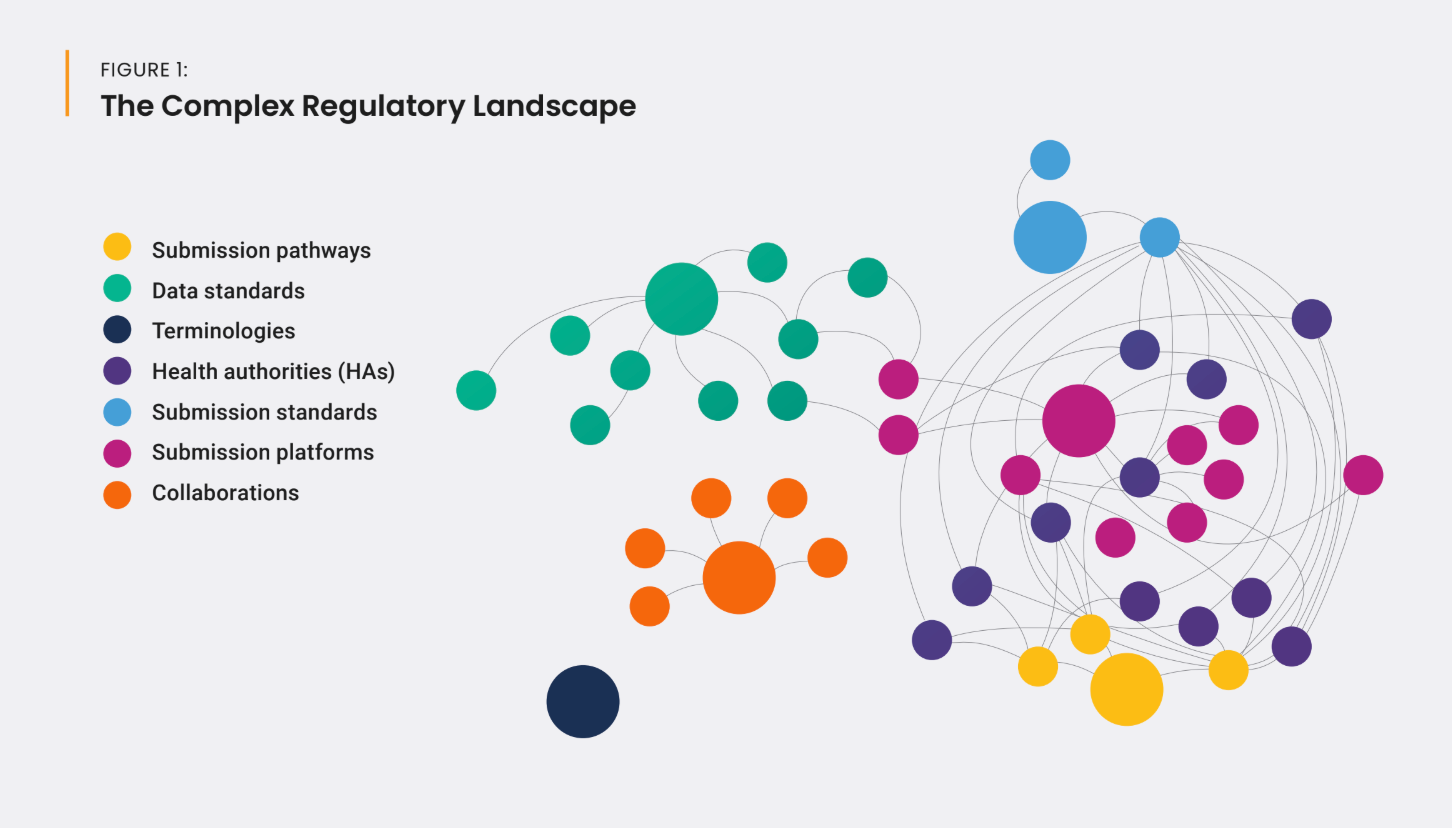
The increasing burden of new, divergent standards and older, outdated standards is hindering innovation; the industry is grappling with “data standards fatigue.” Regulators, standards development organizations (SDOs), sponsors, and solution providers have an opportunity to converge and connect the full ecosystem using existing standards and platforms with global implementations and timelines.
Building the industry framework for convergence and interoperability
Converging standards and regulators with industry and technology requires agreement on harmonized, interoperable data and submissions standards from product development to commercialization and beyond. The industry should embrace existing, functioning, multi- regulator submission platforms that meet the needs of sponsors and HAs. As HAs look increasingly toward real-time and “live” review, global submission formats and platforms are a major source of increasing data exchange efficiency.
Achieving global regulatory convergence and interoperability requires a phased, multi-year strategy with both short- and long-term approaches:
| Short-term strategy | Long-term strategy |
|---|---|
| Only develop new standards with a clear determination of need and global adoption agreements in place | Joint development of unified, fully machine-readable data standards and legal frameworks for mutual reliance and recognition of review outcomes |
| New, interoperable standards must include standard APIs and machine-to-machine information exchange from the start | Collaborative policy development and adoption by HAs, fostered by SDOs, with early input from technology vendors |
| Accelerate progress by adhering to global standards without creating divergent, local implementations | Sustained collaboration toward seamless global exchange of regulatory information, maximizing efficiency for regulators and improving patient access to therapies |
| Leverage existing common electronic submission platforms for rapid, reliable digital data exchange and shared access to review information |
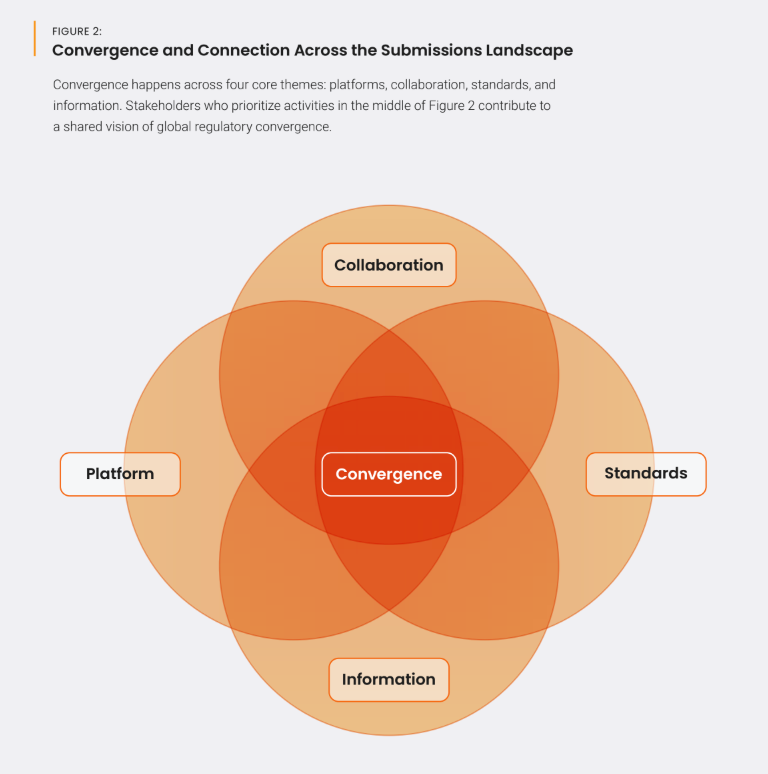
Greater efficiency with a platform approach
Adopt core functionality that serves multiple stakeholders
Technology and digitalization have greatly improved the speed, accuracy, and efficiency of the submissions process, especially in contrast to the days when regulatory filings required box trucks full of submissions paperwork. But, as submissions platforms have multiplied, new challenges have emerged:
- Fragmented systems and data silos prevent visibility and interoperability
- Duplicative data and standards create inefficiency and data quality risks
- Outdated technology and standards slow down processes
Working across multi-stakeholder processes adds even more complexity. Instead of investing in new platforms that may add to these challenges, regulators and industry should consider existing platforms with core functionality that enable faster information exchange, reduce the administrative burden, and lower overall costs. Key capabilities of these platforms should include:
- Bi-directional access and visibility between sponsors and regulatory authorities to respond to questions and resolve discrepancies faster
- Shared access to submissions across HAs that minimizes the need for separate acceptance reviews
- Automated checks for completion, validity, and conformance to standards to reduce validation redundancy
- Shared analysis for faster, transparent, and collaborative review from HAs
- Connected data flows based on global, harmonized data standards to ease data reuse for multiple purposes
- Machine-readable validation to improve compliance and completeness without separate tools and processes
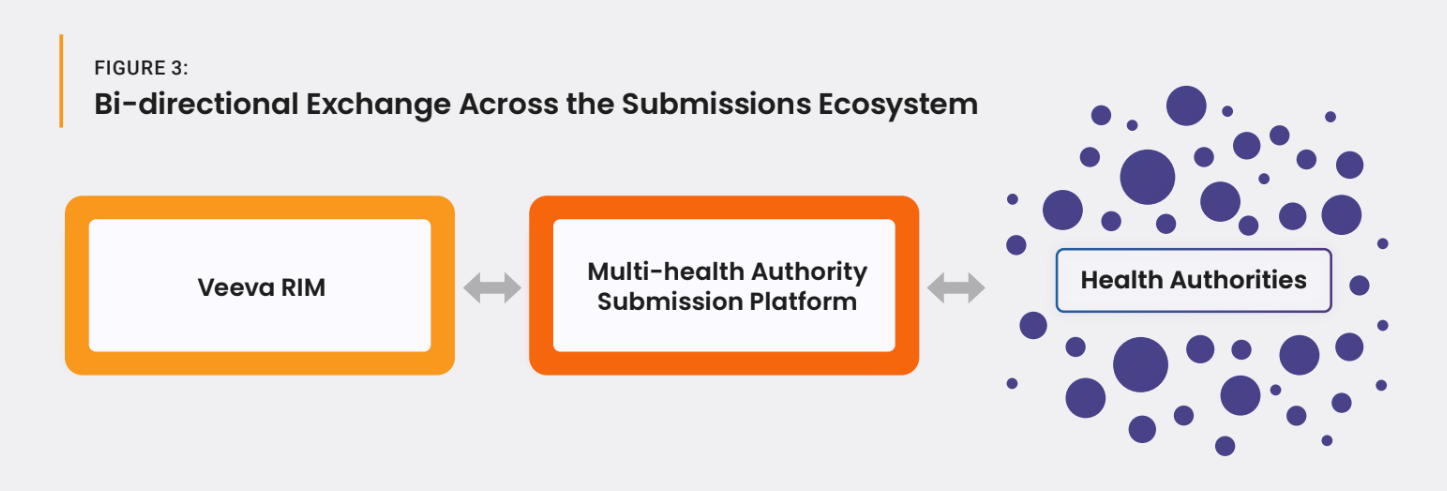
These platforms and benefits exist today and are immediately available. Focusing on existing platforms also reduces time and resources that might otherwise be spent building and piloting new platforms.
Adopting global standards to more easily share information
Simplify and harmonize data models
The duplication of data and submission standards (e.g., eAF, eCTD, IDMP, ePI, SPL, PQ/CMC) creates significant barriers to efficiency. HAs and SDOs should work together to co-create lean, innovative, globally accepted standards. SDOs should drive global acceptance by relying on their charters to resist creating duplicative standards. They can also build digital data exchange into new standards, leaning on all participants to commit to global adoption, shared timelines, and collaborative pilots.
All new standards should support the digital exchange of information and the use of shared submission platforms. These assumptions should be built into new standards from the start. Best practice is to always include an API, machine-readable validation criteria, and commitments from participating HAs to implement within a global adoption timeline.
SDOs should prioritize maximum interoperability during development and engage existing collaborative groups. Including vendors in the entire standards development process increases the technical acuity of standards and improves estimates of implementation effort. In the age of AI, technologists can guide standards development toward data readiness and innovation.
Additionally, the consensus-based standards development process is lengthy and leads to overly-complex standards. SDOs should revisit the consensus model and find ways to increase development speed and simplify final standards. Standards organizations might also consider enabling global adoption and implementation through recommendations for updates to regulatory policy and guidance.
The industry should update existing standards or co-develop new global standards in the form of simplified, harmonized data models and ontologies with fewer elements to exchange information. All new standards should map to global harmonized data models that include the following:
- Readily accessible interoperability information
- Considerations for reducing barriers to access
- Validation and verification requirements based on shared submission platforms
- Global implementation roadmaps with timeline commitments from HAs
Achieving future regulatory efficiency — one characterized by AI-supported review and real-time data exchange — hinges on rethinking fragmented data standards. By streamlining the development process, prioritizing digital readiness, and leveraging the technical expertise of vendors, the global regulatory environment can transition from a slow, paper-based exchange system to a lean, efficient, and innovative digital ecosystem capable of supporting the next century of medical advancement.
Need to collaborate across the industry
Build consensus on standards, use cases, and best practices
Although multi-regulator collaboration platforms and simpler, harmonized, interoperable data standards create the foundation for faster, more efficient, and compliant submissions, stakeholder groups must work together to drive change. And, as the regulatory landscape has grown, so have collaborative groups and supergroups: ICMRA, ISO, HL7, ICH, IPRP, and PIC/S, as well as IRISS, DIA, PhUSE, and RAPS (and many more) work both independently and collaboratively. Although each group exists to help the industry understand and navigate regulations, they also contribute to complexity and fragmentation.
Although advancements in multi-regulator collaboration platforms and the adoption of harmonized data standards lay the groundwork for faster, more compliant submissions, the core challenge is not technological, but organizational. The current growth of stakeholder groups creates a paradox: their collective effort to simplify regulations also contributes to systemic proliferation. Moving forward, these groups and supergroups should shift focus from independent goal-setting to prioritizing interoperability and unified regulatory strategies.
The path to one global submission
The results of these efforts will lead to faster, more efficient, and compliant submissions. The increasing complexity of the regulatory landscape — marked by divergent local requirements and a growing number of stakeholder groups — impedes the delivery of innovative, life-saving medical products to the global public. Advancements in technology and industry collaborations have laid the essential groundwork for more efficient submissions, and efforts are starting to converge. But there is more work to do.
The path to "one global submission" is achievable through collective commitment. Technology, however advanced, can only automate the process that policy dictates. By embracing and improving existing globally- focused digital solutions and harmonizing the policy frameworks that govern them, the life sciences industry can move past "data standards fatigue" and systemic redundancy. This sustained, collaborative action will unlock the speed, efficiency, and patient access that the next generation of medical innovation demands. The blueprint is clear; now, all stakeholders must commit to a unified global strategy to make the vision of faster, safer access to medical innovations a reality.
Learn more about how the industry can converge and connect the global submissions process at Veeva R&D and Quality Summit, Europe.

About the Author
Crystal Allard is senior director, government strategy at Veeva. Prior to Veeva, she spent 15 years driving digital and data modernization in leadership roles at the FDA. Crystal leads Veeva initiatives to modernize and simplify data and submission standards and platforms, engaging with regulators and customers to shape the future of the submissions ecosystem to increase speed to market.
An expert in FDA electronic submissions and health data standards, Crystal contributes to Veeva's product direction and works with the public policy team to position Veeva as a voice in the industry.